Copper »
PDB 3iea-3mn0 »
3mif »
Copper in PDB 3mif: Oxidized (CU2+) Peptidylglycine Alpha-Hydroxylating Monooxygenase (Phm) with Bound Carbon Monooxide (Co)
Enzymatic activity of Oxidized (CU2+) Peptidylglycine Alpha-Hydroxylating Monooxygenase (Phm) with Bound Carbon Monooxide (Co)
All present enzymatic activity of Oxidized (CU2+) Peptidylglycine Alpha-Hydroxylating Monooxygenase (Phm) with Bound Carbon Monooxide (Co):
1.14.17.3;
1.14.17.3;
Protein crystallography data
The structure of Oxidized (CU2+) Peptidylglycine Alpha-Hydroxylating Monooxygenase (Phm) with Bound Carbon Monooxide (Co), PDB code: 3mif
was solved by
X.Siebert,
E.Chufan,
B.A.Eipper,
R.E.Mains,
L.M.Amzel,
with X-Ray Crystallography technique. A brief refinement statistics is given in the table below:
| Resolution Low / High (Å) | 34.63 / 2.00 |
| Space group | P 21 21 21 |
| Cell size a, b, c (Å), α, β, γ (°) | 69.246, 69.715, 83.121, 90.00, 90.00, 90.00 |
| R / Rfree (%) | 20.7 / 23.5 |
Other elements in 3mif:
The structure of Oxidized (CU2+) Peptidylglycine Alpha-Hydroxylating Monooxygenase (Phm) with Bound Carbon Monooxide (Co) also contains other interesting chemical elements:
| Nickel | (Ni) | 1 atom |
Copper Binding Sites:
The binding sites of Copper atom in the Oxidized (CU2+) Peptidylglycine Alpha-Hydroxylating Monooxygenase (Phm) with Bound Carbon Monooxide (Co)
(pdb code 3mif). This binding sites where shown within
5.0 Angstroms radius around Copper atom.
In total 2 binding sites of Copper where determined in the Oxidized (CU2+) Peptidylglycine Alpha-Hydroxylating Monooxygenase (Phm) with Bound Carbon Monooxide (Co), PDB code: 3mif:
Jump to Copper binding site number: 1; 2;
In total 2 binding sites of Copper where determined in the Oxidized (CU2+) Peptidylglycine Alpha-Hydroxylating Monooxygenase (Phm) with Bound Carbon Monooxide (Co), PDB code: 3mif:
Jump to Copper binding site number: 1; 2;
Copper binding site 1 out of 2 in 3mif
Go back to
Copper binding site 1 out
of 2 in the Oxidized (CU2+) Peptidylglycine Alpha-Hydroxylating Monooxygenase (Phm) with Bound Carbon Monooxide (Co)
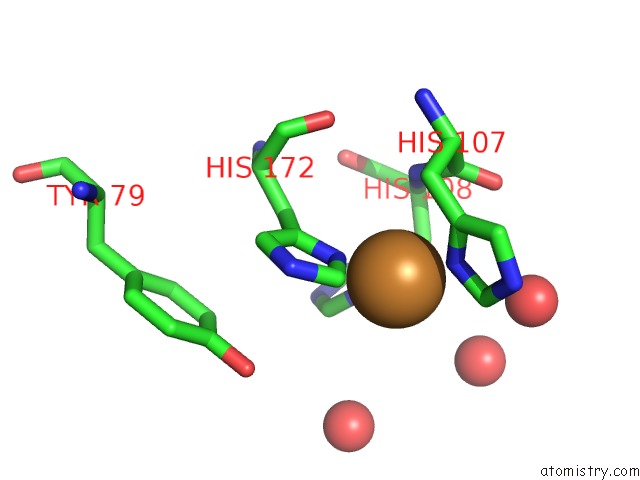
Mono view
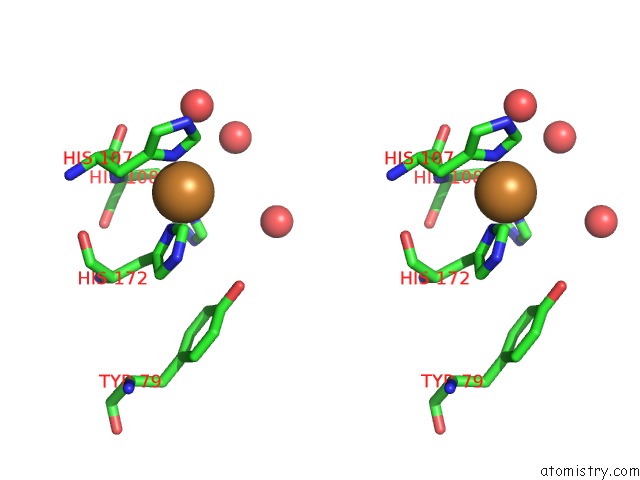
Stereo pair view
Mono view
Stereo pair view
A full contact list of Copper with other atoms in the Cu binding
site number 1 of Oxidized (CU2+) Peptidylglycine Alpha-Hydroxylating Monooxygenase (Phm) with Bound Carbon Monooxide (Co) within 5.0Å range:
|
Copper binding site 2 out of 2 in 3mif
Go back to
Copper binding site 2 out
of 2 in the Oxidized (CU2+) Peptidylglycine Alpha-Hydroxylating Monooxygenase (Phm) with Bound Carbon Monooxide (Co)
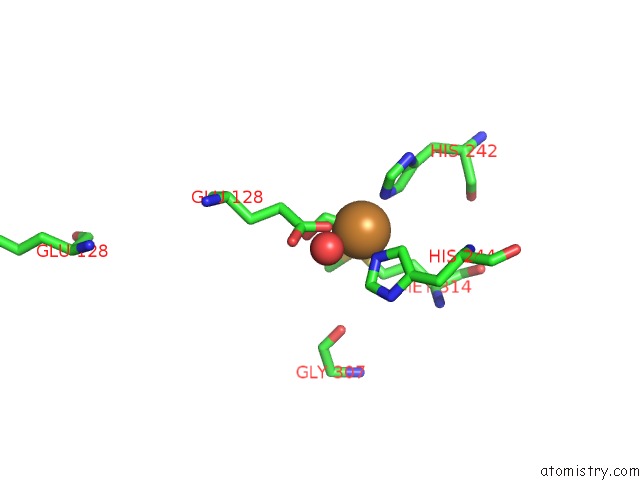
Mono view
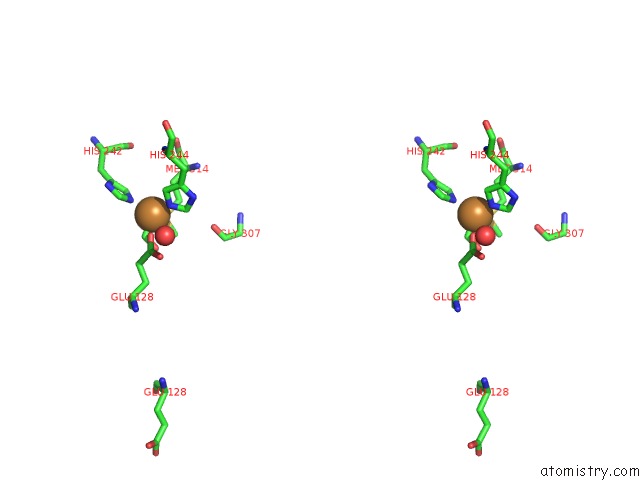
Stereo pair view
Mono view
Stereo pair view
A full contact list of Copper with other atoms in the Cu binding
site number 2 of Oxidized (CU2+) Peptidylglycine Alpha-Hydroxylating Monooxygenase (Phm) with Bound Carbon Monooxide (Co) within 5.0Å range:
|
Reference:
E.E.Chufan,
S.T.Prigge,
X.Siebert,
B.A.Eipper,
R.E.Mains,
L.M.Amzel.
Differential Reactivity Between the Two Copper Sites of Peptidylglycine Alpha-Hydroxylating Monooxygenase (Phm) J.Am.Chem.Soc. V. 132 15565 2010.
ISSN: ISSN 0002-7863
PubMed: 20958070
DOI: 10.1021/JA103117R
Page generated: Wed Jul 31 01:19:21 2024
ISSN: ISSN 0002-7863
PubMed: 20958070
DOI: 10.1021/JA103117R
Last articles
Zn in 9MJ5Zn in 9HNW
Zn in 9G0L
Zn in 9FNE
Zn in 9DZN
Zn in 9E0I
Zn in 9D32
Zn in 9DAK
Zn in 8ZXC
Zn in 8ZUF